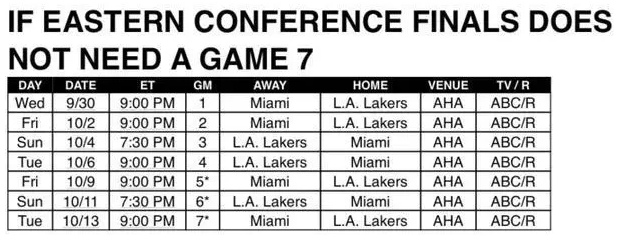
| 成都生物所开发新型生物信息学软件 |
近年来,随着测序技术的快速发展,动物(尤其是非模式动物)全基因组数据(WGS)获取变得简单、便宜,这使得数据库中全基因组数据量呈指数态势增长。全基因组测序数据往往同时包含线粒体序列和核基因序列,通过生物信息学的方法从中提取并组装完整的线粒体基因组,是非常好的技术手段。虽然已有部分学者开发了一些生物信息学软件用以实现此目的,但是,其大都具有自身的局限性,包括需要人工提供参考序列(“种子”)、数据兼容性差、准确率低等难点,未能高效、准确、便捷地完成相关任务。为克服这一难点,提升研究人员的工作效率,近日,中国科学院成都生物研究所(以下简称成都生物所)李家堂课题组开发了一种不依赖“种子”序列从动物全基因组数据中提取并组装线粒体全基因组的方法,并使用Python及C++编写为软件——MEANGS。
据介绍,MEANGS工作流程比较简单,极大地提升了研究人员的可操作性。首先,利用预置的线粒体模块数据库,MEANGS使用nhmmer根据线粒体序列特征对输入的二代数据进行预筛选并获取潜在的线粒体编码reads;获得潜在的线粒体编码reads后,MEANGS利用线性迭代算法(SSAKE)(C++改写)组装模块对reads进行组装获取线粒体编码contigs;相关contigs经由nhmmer进行二次筛选,非冗余线粒体编码contigs将被选择作为“种子”序列用于全线粒体基因组的组装;最后利用“种子”序列,MEANGS再次使用组装模块以“种子”contigs为基础,组装完整的线粒体基因组。另外,针对组装好的线粒体基因组,MEANGS可对编码基因进行辅助注释。

图1 MEANGS利用动物全基因组数据组装线粒体基因组的工作流程 成都生物所提供
针对MEANGS,本研究使用了来自不同类群(哺乳类、鸟类、鱼类、爬行类、两栖类,昆虫和软体动物),不同测序质量的16组数据来进行相关测试。同时,本研究选取了其他三款同类型软件(norgal, NOVOPlasty, MitoZ)进行同条件测试,并基于常规的评价标准(运行时长、内存使用、准确性、完整性、完成数量等)对软件的表现进行评价。测试及比较结果表明,MEANGS数据兼容性最好,完成了所有测试数据的测试,而其他三款软件均有失败案例。在常规的评价指标上,MEANGS表现出最优的整体性能,除了在内存使用上稍逊色于norgal,其余表现均优于其他软件。MEANGS的开发有利于研究人员开展相关工作,推动相关学科的高质量快速发展。

图2 MEANGS与其他三款软件相比表现出最好的数据兼容性 成都生物所提供

图3 MEANGS与其他三款软件在常规评价指标上显示出明显优势 成都生物所提供
该研究成果以MEANGS: an efficient seed-free tool for de novo assembling animal mitochondrial genome using whole genome NGS data为题,已在生物信息学国际顶级期刊Briefings in Bioinformatics(IF: 11.62)在线发表。成都生物所在读博士研究生宋梦洹,特别研究助理严超超博士为论文的共同第一作者,李家堂研究员为论文的通讯作者。本研究得到成都生物所高级工程师蒋海波博士和硕士生桂承波的大力帮助。本研究得到中国科学院B类先导科技专项、科技部第二次青藏高原综合科学考察研究项目、中国科学院对外合作重点项目、中国科学院西部之光交叉团队项目、中国科学院前沿科学重点研究项目及四川省杰出青年科技人才项目等项目的资助。
相关论文链接:
https://doi.org/10.1093/bib/bbab538
近年来,随着测序技术的快速发展,动物(尤其是非模式动物)全基因组数据(WGS)获取变得简单、便宜,这使得数据库中全基因组数据量呈指数态势增长。全基因组测序数据往往同时包含线粒体序列和核基因序列,通过生物信息学的方法从中提取并组装完整的线粒体基因组,是非常好的技术手段。虽然已有部分学者开发了一些生物信息学软件用以实现此目的,但是,其大都具有自身的局限性,包括需要人工提供参考序列(“种子”)、数据兼容性差、准确率低等难点,未能高效、准确、便捷地完成相关任务。为克服这一难点,提升研究人员的工作效率,近日,中国科学院成都生物研究所(以下简称成都生物所)李家堂课题组开发了一种不依赖“种子”序列从动物全基因组数据中提取并组装线粒体全基因组的方法,并使用Python及C++编写为软件——MEANGS。
据介绍,MEANGS工作流程比较简单,极大地提升了研究人员的可操作性。首先,利用预置的线粒体模块数据库,MEANGS使用nhmmer根据线粒体序列特征对输入的二代数据进行预筛选并获取潜在的线粒体编码reads;获得潜在的线粒体编码reads后,MEANGS利用线性迭代算法(SSAKE)(C++改写)组装模块对reads进行组装获取线粒体编码contigs;相关contigs经由nhmmer进行二次筛选,非冗余线粒体编码contigs将被选择作为“种子”序列用于全线粒体基因组的组装;最后利用“种子”序列,MEANGS再次使用组装模块以“种子”contigs为基础,组装完整的线粒体基因组。另外,针对组装好的线粒体基因组,MEANGS可对编码基因进行辅助注释。
图1 MEANGS利用动物全基因组数据组装线粒体基因组的工作流程 成都生物所提供
针对MEANGS,本研究使用了来自不同类群(哺乳类、鸟类、鱼类、爬行类、两栖类,昆虫和软体动物),不同测序质量的16组数据来进行相关测试。同时,本研究选取了其他三款同类型软件(norgal, NOVOPlasty, MitoZ)进行同条件测试,并基于常规的评价标准(运行时长、内存使用、准确性、完整性、完成数量等)对软件的表现进行评价。测试及比较结果表明,MEANGS数据兼容性最好,完成了所有测试数据的测试,而其他三款软件均有失败案例。在常规的评价指标上,MEANGS表现出最优的整体性能,除了在内存使用上稍逊色于norgal,其余表现均优于其他软件。MEANGS的开发有利于研究人员开展相关工作,推动相关学科的高质量快速发展。
图2 MEANGS与其他三款软件相比表现出最好的数据兼容性 成都生物所提供
图3 MEANGS与其他三款软件在常规评价指标上显示出明显优势 成都生物所提供
该研究成果以MEANGS: an efficient seed-free tool for de novo assembling animal mitochondrial genome using whole genome NGS data为题,已在生物信息学国际顶级期刊Briefings in Bioinformatics(IF: 11.62)在线发表。成都生物所在读博士研究生宋梦洹,特别研究助理严超超博士为论文的共同第一作者,李家堂研究员为论文的通讯作者。本研究得到成都生物所高级工程师蒋海波博士和硕士生桂承波的大力帮助。本研究得到中国科学院B类先导科技专项、科技部第二次青藏高原综合科学考察研究项目、中国科学院对外合作重点项目、中国科学院西部之光交叉团队项目、中国科学院前沿科学重点研究项目及四川省杰出青年科技人才项目等项目的资助。
相关论文链接:
https://doi.org/10.1093/bib/bbab538
热门